No more cloning: quick and easy CRISPR with sgRNA gene fragments
Editor’s note: This post was contributed by Ralitsa Madsen, a PhD student at the University of Cambridge. Below she shares her journey in harnessing the power of CRISPR. Ultimately, she was able to create point mutation knockins in HEK293s using CRISPR with up to 18% efficiency without selection. Check out this post for her tips and tricks!
Want to be Benchling’s guest blogger? Contact us.
Introduction
When I started my PhD project in October 2015, I knew three things for sure:
My project involved modelling disease-specific point mutations in human pluripotent stem cells.
I would generate these point mutations using CRISPR/Cas9, a technology providing scientists with state-of-the-art molecular DNA scissors.
I had never used CRISPR/Cas9 and did not have a clue what conditions would work best in my hands!
It is now April 2016, and I have come a long way. Following multiple rounds of optimisations, trials-and-errors and long working hours, I have become a confident user of this technology. However, this progress would have been impossible had it not been for the multitude of protocols that have been published across scientific journals and blogs around the web. It is time for another blog post of this kind, now with me in the writer’s seat, taking you through my CRISPR journey.
Optimize conditions in HEK293s cells
Anyone hoping to use CRISPR to establish a reliable cellular model of a specific human disease should have as their top priority the mitigation of off-target effects. Although wildtype Cas9 off-target effects in human pluripotent stem cells appear to be very low, I decided to be on the safest side possible and started off by designing single guide RNAs (sgRNA) for the less promiscuous Cas9 nickase system, focusing on only one of the three planned point mutations.
I cloned each individual pair into the Cas9 nickase PX462 plasmid (Addgene #48131 for V1.0 and #62988 for V2.0) and co-nucleofected 1.5 ug of each construct together with 200 pmol 150-bp-long homology-directed repair (HDR) template into 1,000,000 HEK293s cells (more on HDR design here). To screen for successful knockins, I used a quick and easy restriction fragment length polymorphism (RFLP) assay.
Tip 1: Use an easy and cheap cell culture system to optimize your CRISPR experiments. It saves you money and time to perform your CRISPR optimisations in culture systems with robust and high transfection efficiency, such as HEK293 cell line. It is best to move forward to more demanding and expensive cultures once you have ensured that your reagents are working.
Tip 2: You might be able to improve knock-in efficiency with asymmetric HDR template design. In conventional HDR template design, you put the point mutation in the middle and design 45-75 bp homology arms on both sides of the mutation. Richardson et al. showed that the knock-in efficiency can be higher when you design the HDR template complementary to the strand of sgRNA and with a 36-bp homology arm at 3’ of PAM and 91-bp arm at 5’ of PAM1.
Tip 3: Introduce a mutation at the PAM site to prevent Ca9 from degrading the HDR template. If possible, introduce a silent mutation at the PAM site to prevent repeated cutting of a successfully targeted genomic region. However, changing the PAM from NGG to NAG will not do the trick, as NAG works as a cryptic PAM site for Cas92. If it is not possible to mutate the PAM, introduce 2-3 silent mutations in the sgRNA-specific PAM-proximal region. For more on how to do this using Benchling, click here.
Tip 4: Introduce a novel restriction enzyme site when designing the HDR template. Ensure that one of the silent mutations introduces a novel restriction enzyme site, allowing you to identify the presence of knockins across the cell population (no single-cell cloning!) by a simple restriction fragment length polymorphism (RFLP) assay (< 5h hands-on work). This online tool will help you design novel restriction enzyme sites.
Tip 5: There are multiple assays to test sgRNA cleavage efficiency. In addition to RFLP assay, there are other commercially available assays to test sgRNA cleavage efficiencies in vitro, such as Surveyor Mismatch Cleavage Assays.
What didn’t work in my optimization journey
My initial efforts led me to a blind alley. Despite tweaking different parameters, I still had a hard time obtaining a knockin efficiency above 5 % for the particular gene and point mutation without any enrichment. I tested multiple sgRNAs: one of the pairs did not work at all, the remaining two gave positive results, with one pair being twice as efficient. This is consistent with reports that even overlapping sgRNAs can exhibit markedly different efficiencies.
I also tried to extend the time to collection, but found that culturing the cells for longer than 48h post-nucleofection had little to no positive effect on the observed knockin efficiency. Having read that the Cas9 nickase is less efficient than its wildtype counterpart, I then cloned my sgRNAs into the wildtype Cas9 PX459 vector (Addgene #48139 for V1.0 and #62988 for V2.0) ; this proved to be another cul-de-sac as the knockin efficiency once again happened to be low.
You might wonder why I was not happy with 5%? After all, this is a bulk cell population, and enrichment by single-cell cloning should save me, right? Yes, in HEK293s cells. However, it is common to observe a higher knockin efficiency in immortalised cell lines with more promiscuous DNA repair pathways3. Thus, I worried that a relatively low efficiency in HEK293s would mean “below detection limit” in pluripotent stem cells.
Optimization that works: gBlocks encoding FE-modified sgRNAs
Fortunately, a Stem Cell Reports Resource paper came to my rescue just at the right time4. Mandana Arbab and her colleagues reported the development of a cloning-free CRISPR protocol relying on gBlocks (gene fragments) encoding FE-modified sgRNAs. FE stands for an A-U flip and a stem extension (Figure 1). This modifications developed by Chen et al. promote enhanced stability5. As a result, the sgRNAs linger in the cell for longer.
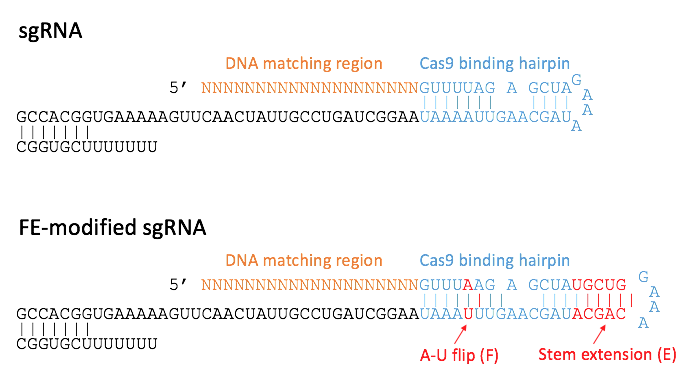
The gBlocks protocol seemed straight-forward, and I was tempted by a method that obviates the need for plasmid-based cloning of every single sgRNA to be tested. This could save me precious time when preparing the reagents for the remaining two point mutations. With helpful advice from Mandana herself, I took the turn and gave the gBlocks a go.
The results were hard to believe; the wildtype Cas9 achieved efficiencies close to 18 % (n=3) and although less impressive, the Cas9 nickase also homed in on roughly 8 %. This increase in efficiency was not due to the fact that the gBlocks protocol required 5 ug Cas9-expressing plasmid, as a side-by-side comparison of my previous method ramping up the amount of Cas9 plasmid remained half as efficient as the gBlocks. Thus, it seems that the enhanced stability of the FE-modified sgRNAs are key to the success of the method.
Subsequently, I have generated the reagents for the remaining two point mutations, and although the knockin efficiencies vary greatly with DNA location, the rapidity and easiness of the gBlocks protocol has made it my gold-standard CRISPR approach.
You can find the detailed protocol here.
Discussion: how about off-target effects?
You may ask if I have completely forgotten about off-target effects, especially when ramping up to 5 ug wildtype Cas9 plasmid? Not at all. I tried to address this issue by testing the recently published eSpCas9; an engineered Cas9 version with minimal off-target effects. Unfortunately, the previous efficiency of 18 % with wildtype Cas9 dropped below 2 % when switching to eSpCas9.
After some troubleshooting and CRISPR Google groups mining, I found out that the eSpCas9 is incompatible with the extra 5’G appended to sgRNAs that do not start with a G, a common modification to ensure transcription downstream of a U6 promoter. Therefore, a switch to eSpCas9 is not a viable option for me.
Instead, I am currently in the process of testing a recombinant Cas9 protein with the gBlocks methods. Due to the proteins relatively short life within the cell, off-target effects are lower compared to the plasmid-based method. In addition, plasmid-based method (double-stranded) further carries a risk of random integration into the genome compared to gBlocks methods (single-stranded). In any case, I will ultimately rely on thorough screening methods to ensure that the final engineered stem cells are free of unwanted mutations.
Tip 5: eSpCas9 is sensitive to sgRNAs supplemented with an extra G at the start.
Flowchart of my CRISPR journey
Finally, here is my step-by-step CRISPR optimisation journey – good luck with yours!
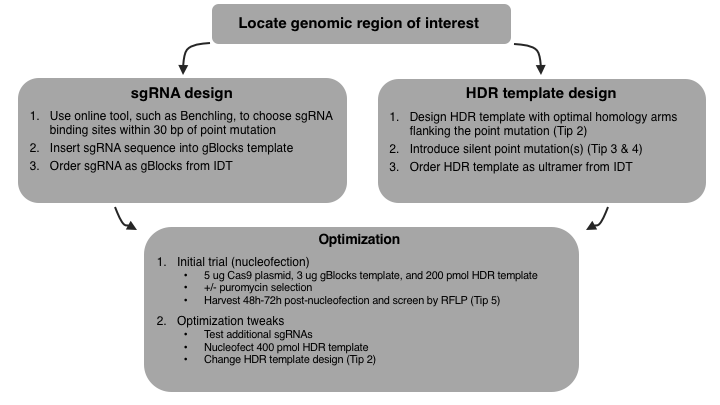
Tip 6: Benchling has wizards to help you easily design sgRNA and HDR template. If you want to learn more about designing sgRNA and HDR template, you might also like the posts “How to Design gRNAs to Target Your Favorite Gene” and “How to Design Homologous Recombination Template for CRISPR”.
Conclusion
Using FE-modified sgRNA increased my knock-in efficiency up to 18% in HEK293s without selection. If you want to give this method a shot, you can use my protocol for synthesizing FE-modified sgRNA with gBlocks template.
Finally, follow Benchling on Twitter or Facebook so you don’t miss future posts!
References
Richardson, Christopher D et al. "Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA." Nature biotechnology (2016).
Zhang, Yilan et al. "Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells." Scientific reports 4 (2014).
Hendriks, William T, Curtis R Warren, and Chad A Cowan. "Genome Editing in Human Pluripotent Stem Cells: Approaches, Pitfalls, and Solutions." Cell stem cell 18.1 (2016): 53-65.
Arbab, Mandana et al. "Cloning-free CRISPR." Stem cell reports 5.5 (2015): 908-917
Chen, Baohui et al. "Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system." Cell 155.7 (2013): 1479-1491.
Slaymaker, Ian M et al. "Rationally engineered Cas9 nucleases with improved specificity." Science 351.6268 (2016): 84-88.